Rolling friction at a microscopic scale is studied with the help of a simple two-dimensional model. MD simulations show that rolling of spherical lubricant molecules exists only for concentrations lower than the concentration of the close-packed monolayer. At concentrations higher than a critical one, the rolling of nearest neighboring molecules is hindered because of jamming of lubricant molecules. It exists an optimal concentration which provides the minimum of kinetic friction. Methods how to avoid jamming and increase the range of operation of the rolling mechanism of friction are discussed.
As is known, coefficients of rolling friction are generally 102 to 103 times lower than those of sliding friction for corresponding materials. The main source of friction in macroscopic rolling is dissipation of energy involved in deformation of the objects. In this context the following intriguing question emerges: may a similar mechanism work at a microscopic scale, i.e., may a ball-shape molecules such as almost spherical C60 molecules (fullerenes) work as a "molecular bearing"? It could be very promising for realization in nano- and micromachines. Unfortunately, these anticipations have not been confirmed at experiments: the lowest friction coefficient between two C60 films was found to be of order μ ~ 0.15.
Our goal is to understand what physics may yield the lowest friction coefficient attainable for molecular rolling and which system parameters might provide it. First, we show that the microscopic rolling is hindered because of jamming of lubricant molecules. Second, we find that nanobearings might indeed work as well as macroscopic ones, but one has to choose properly the macroscopic counterpart, which here turns out to be a perfect rack-and-pinion matching as in cogwheels.
 We use MD simulation for a 2D system,
where all atoms have the unit mass (m = 1) and
interact via the 6-12 Lennard-Jones (LJ) potential
VLJ(r) = V0 [(R0/r)12
− 2(R0/r)6]
with the amplitude V0=1 and the equilibrium distance R0=1 (this defines our dimensionless system of units).
We use MD simulation for a 2D system,
where all atoms have the unit mass (m = 1) and
interact via the 6-12 Lennard-Jones (LJ) potential
VLJ(r) = V0 [(R0/r)12
− 2(R0/r)6]
with the amplitude V0=1 and the equilibrium distance R0=1 (this defines our dimensionless system of units).
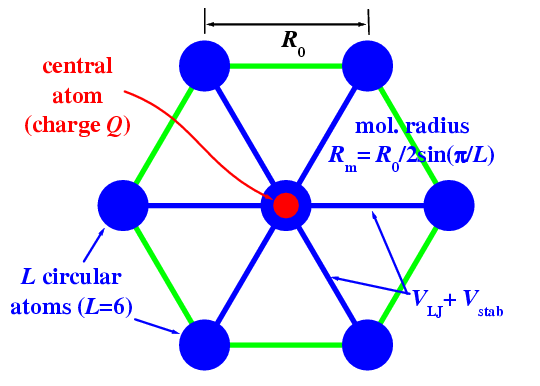
A single circular molecule. Each molecule is constructed of L+1 atoms (see figure to the right): one central atom and L atoms on circle of radius Rm=R0/2 sin(p/L) so that the equilibrium distance between the circular atoms is R0. The circular atoms of a given molecule are coupled with the central atom, additionally to the LJ potential, by stiff springs of the elastic constant Km, Vstab(r)=1/2 Km(r−Rstab)2, where the distance Rstab= Rm + (12 V0/KmRm)[(R0/Rm)6 − (R0/Rm)12] is chosen so that the total potential VLJ(r) + Vstab(r) has the minimum at r=Rm.
The 2D system. We put N lubricant molecules between the two (bottom and top) substrates.
To reduce the number of parameters, we model each substrate
by a closely-packed monolayer constructed of the same molecules and
pinned to the corresponding (top or bottom) substrates.
 In the 2D model, the closely-packed monolayer corresponds to
a chain of length M with the periodic boundary condition
along the x direction as shown (figure to the right).
The two outmost atoms of each substrate's molecule are coupled to the "pinning sites"
(shown by red circles in figure) with the help of
stiff springs of the elastic constant Ks, Vsub(r)=1/2Ksr2.
These "pinning sites" (PS) itself constitute a rigid lattice which models the
removed part of the semi-infinitive substrate.
In the 2D model, the closely-packed monolayer corresponds to
a chain of length M with the periodic boundary condition
along the x direction as shown (figure to the right).
The two outmost atoms of each substrate's molecule are coupled to the "pinning sites"
(shown by red circles in figure) with the help of
stiff springs of the elastic constant Ks, Vsub(r)=1/2Ksr2.
These "pinning sites" (PS) itself constitute a rigid lattice which models the
removed part of the semi-infinitive substrate.
The bottom PS lattice is fixed at x=y=0, while the top
substrate may move
in two dimensions x and y.
A load force fload is applied to the top PS lattice
(in what follows all forces are presented in units "force per one pinned molecule").
Also, the top PS lattice is connected by
a weak spring of elastic constant kspring with a base which
moves with a constant velocity vdrive.
The spring force, which corresponds to the frictional force, is monitored
during simulation.
For the elastic constants we choose Km= Ks= 100 and kspring= 10
Because a 2D model cannot reproduce even qualitatively the true phonon spectrum of a 3D system, we use Langevin equation of motion with Gaussian random force corresponded to a given temperature T, and the damping force fh,x= −mη(y) dx/dt − mη(Y−y) (dx/dt − dX/dt), where x, y are the atomic coordinates and X, Y are the coordinates of the top PS lattice (the force fh,y is defined in the same way). The viscous damping coefficient decreases exponentially with the distance from the corresponding PS lattice, η(y) = η0 [1 − tanh(y/2Rm)], where we typically used η0=1.

For loads fload≥ 0.5 the Amontons law operates in the smooth-rolling regime. Note that the rolling is not frictionless, i.e., the static friction is nonzero, μs > 0.15. In order to rotate a lubricant molecule over the substrate, one has to break at least one interatomic bond. Let V0' be the total energy of these bonds (V0' ~ V0). Thus, one has to apply the driving force f0' ~ 2V0'/R0 in order to start rolling, where the factor 2 emerges because of two substrates. Then, the Amontons law works owing to the proportionality V0' ∝ fload' (here fload' is the local load force), which follows from the relationship ∂VLJ/∂y ~ ∂VLJ/∂x.
When the driving velocity increases, the system exhibits a transition from stick-slip to smooth motion, where slips (in the stick-slip regime) as well as the smooth motion correspond to rolling of lubricant molecules.
Example: θ = 2/3 (N = 20, M = 30), T = 0, Q = 0, fload= 2
vdrive=1: stick-slip (sticks due to jamming) ||| vdrive=1.5: smooth rolling
(animated gif 3 Mb) ||| (animated gif 1.1 Mb)

Dependence of the kinetic friction on the lubricant concentration (vdrive= 2.25, T=0, Q=0, M=30; θoptimum~ 0.3)
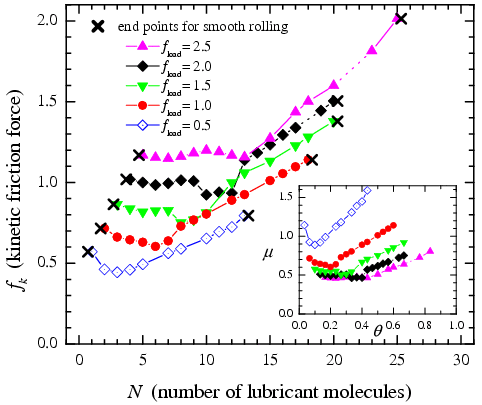
Figure: At very low concentrations the smooth rolling is destroyed and changes to stick-slip or creep motion because of strong increase of the local load fload' and, therefore, the local driving f0'. The range of concentrations where the smooth rolling exists is indicated by crosses in the Figure. The most important result is that the smooth rolling is also destroyed at high concentrations because of jamming of the lubricant. Jamming, or the "traffic-jam" effect in driven systems is well known. In the case of rolling friction the jamming is, however, much more dramatic than in the case of sliding friction with conventional lubricants. When two nearest neighboring (NN) rolling molecules come in close contact, they hinder mutual rolling, because the two sides of colliding molecules roll in opposite directions. In the result, both molecules stop to roll. Then the jam grows in size and totally destroys the smooth rolling regime. Thus, the smooth rolling cannot exist at high concentration of the lubricant, e.g., for the closely packed layer Θ=1.
reduce concentration
increase temperature (to break intermolecular bonds)
avoid collisions (e.g., Q > 0)
 |
Figure: Dependence of the kinetic friction
(a) on the temperature T for Q=0, and
(b) on the repulsion amplitude Q for T=0 |
 |
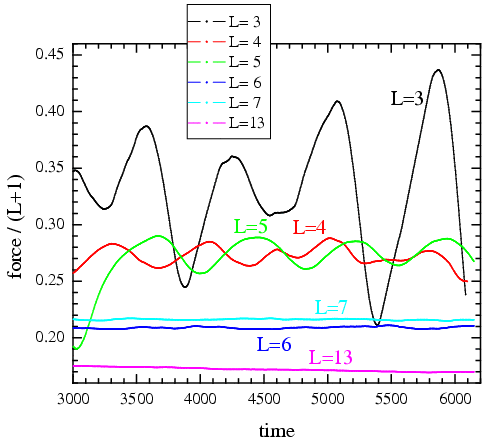
Figure: Decrease of the critical velocity of the transition from stick-slip to smooth rolling (θ=2/3, fload=2) |
(b) θ=2/3 (N=20, M=30), fload=2, T=0.5, Q=2, vdrive=0.2 (animated gif 7.6 Mb)
Role of the shape (size) of the rolling lubricant molecules (Figure for θ=2/3, fload= 2, vdrive= 2.25): a rounder wheels roll better!
 |
Movies:
|
Even without jams, the friction with spherical molecules is still quite large, of order μ > 0.1, like with conventional oil-based lubricants. Above, however, we used deformable substrates constructed of the same spherical molecules. One may expect that the using of more rigid substrates can improve the situation. Indeed, let us consider two rigid substrates with spherical lubricant molecules in between. The substrates, pressed together by a load force Fl= Nsfl, consist of rigid atomic chains of length Ns and lattice constant Rs, so that the system size in the sliding direction is Lx= NsRs and the total mass of the substrate is Nsms. The bottom rigid substrate is fixed at x=y=0, the top one is free to move in both x and y, while driven along x through a spring of elastic constant ks moving with speed vs. The spring force F, whose maximum value before the onset of sliding measures the static friction force Fs, and whose average during smooth motion Fk=‹F› is the kinetic friction force, is monitored during simulation. Between the substrates we place circular lubricant molecules. As above, each molecule consists of one central atom and of L atoms on circle of radius Rm= Rll/2 sin(π/L) so that their chord distance is Rll=R0. All atoms interact via a 12-6 Lennard-Jones (LJ) potential VLJ (r) = Vαα' [(Rαα'/r)12 − 2(Rαα'/r )6], where α,α' = "s" or "l" for the substrate or molecular atoms respectively. The latter are additionally coupled to the central atom by stiff springs of the elastic constant Km=100. Thus, the lubricant-lubricant interaction is described by the parameters Vll and Rll, while the lubricant-substrate interaction, by Vsl and Rsl (direct interaction between the top and bottom substrates is omitted). As above, we use dimensionless units, where ms=ml=1, Rll=1, and the energy parameters Vαα' takes values around Vαα' ~1. Simulations were done for molecule sizes from L=5 (the simplest circular molecule) up to L=13 and 14, which may be considered as a 2D version of fullerene. In fact in the 3D case, the surface area of the spherical molecule is s = 4πRm2. With L3=60 atoms on the surface, this gives s ≈ L3Rll2, or Rm/Rll≈ 2.18. In 2D, the length of the circle is 2πRm≈ LRll, or L ≈ 2πRm/Rll, which leads to L ≈ 13.7 for the same ratio Rm/Rll as for 3D fullerenes.
A single rigid molecule. We first consider a rigid molecule, Vll= ∞ and Km= ∞, fix X of the top substrate and seek the potential energy minimum V by varying its y-coordinate Y, the molecular center (xc, yc) and the rotation angle φ. The X dependence of V, (xc, yc), and φ defines the adiabatic trajectory, which describes the joint substrate and lubricant motion when infinitely slow. We extract the activation energy Ea= max[V(X)] − min[V(X)], and the static friction force, approximated as fs= max[ dV(X)/dX ] (fs~ Ea in our units).
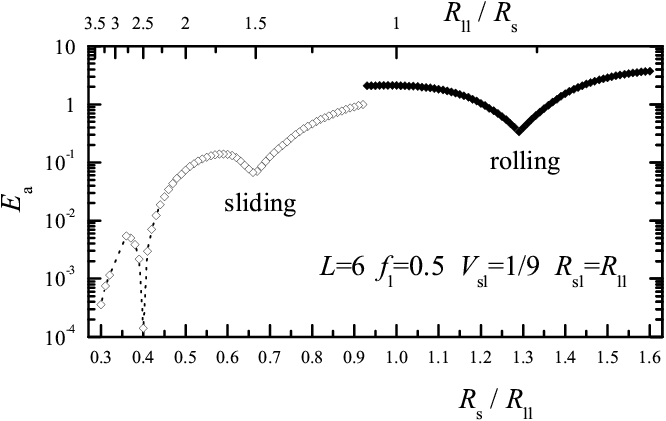 |
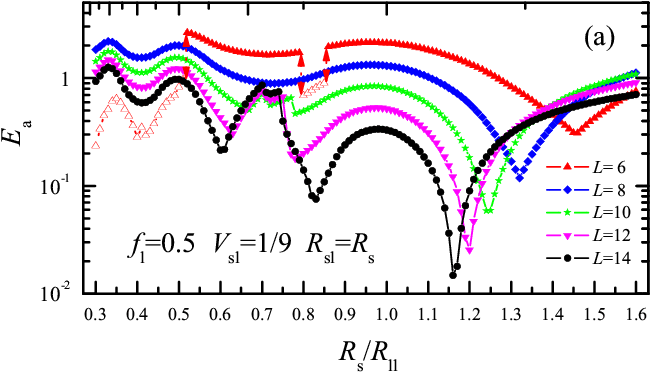
Figure 1: Activation energy Ea as a function of the substrate lattice constant Rs for the rigid L=6 molecule, for fl= 0.5, Vsl= 1/9, and Rsl= Rll. Open symbols correspond to sliding, solid symbols to rolling |
 |
 |
|
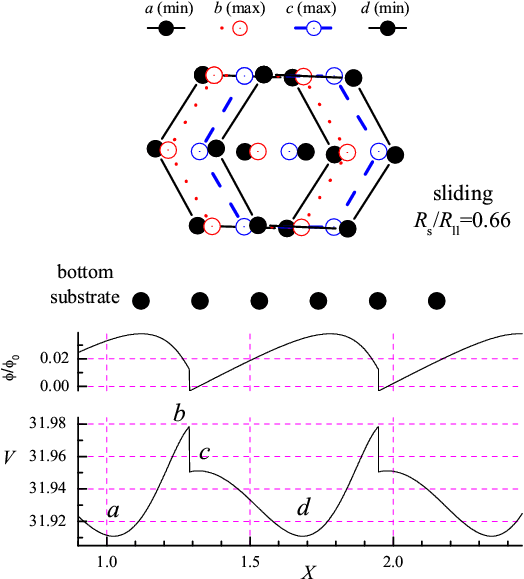
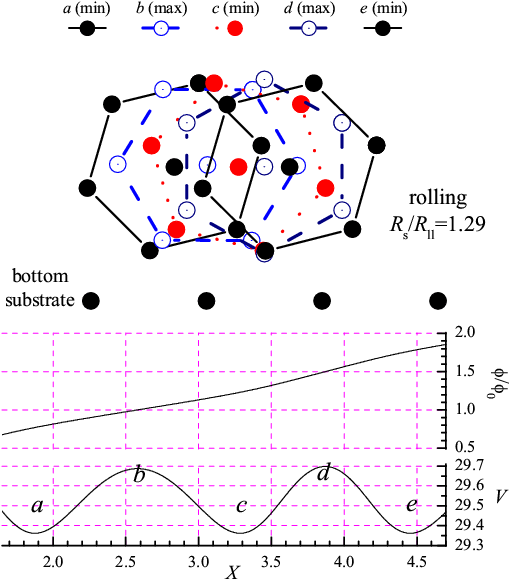
Figure 2: Sliding adiabatic motion of the rigid L=6 molecule for Rs/Rll= 0.66 (Δφ < φ0, left panel) and rolling for Rs/Rll= 1.29 (Δφ > φ0, right panel). Other parameters as in Fig.1. Lower panels: X-dependence of the potential energy V(X), and the angle φ(X)/φ0. Top panel: configurations as the molecule moves from one minimum of V(X) to the next. |
|
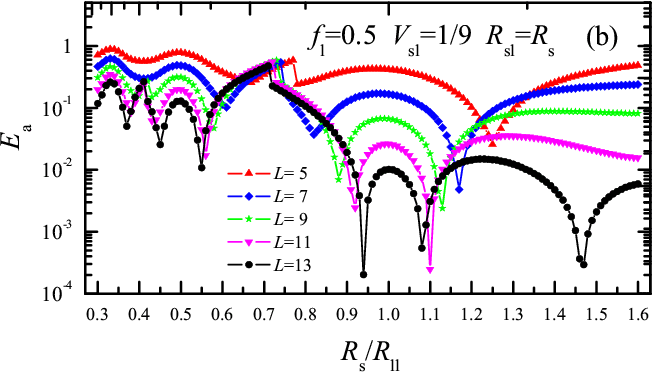
Varying Rs in Fig.1 at fixed value of Rsl for the lubricant-substrate interaction, we find that for Rs< Rll rolling replaces sliding already around Rs/Rll= 2/3. Preference for rolling over sliding increases for increasing load fl and for decreasing interaction strength Vsl (choosing alternatively Rsl= Rs rolling is even more prevalent than for fixed Rsl). When sliding wins over rolling, it generally provides a lower activation energy. Recalling that φ0= 2π/L, the region of parameters for rolling should increase with L − a rounder wheel rolls better! The Rs dependence of Ea(Rs) for increasing size L (see Fig.3) shows rolling for all Rs and for all L≥5, except for L=6 which shows both rolling and sliding (see open symbols in Fig.3a). As Rs varies, the value of Ea oscillates by more than two orders of magnitude for even L and more than three for odd L, with deep sharp minima separated by broad maxima.
 |
 |
|
Figure 3: Rigid molecule activation energy Ea versus Rs/Rll for fl= 0.5 and Vsl= 1/9. Unlike Fig.1, here Rsl= Rs. Panel (a): even L=6, 8, 10, 12, 14; panel (b), odd L=5, 7, 9, 11, 13. Empty triangles in L=6 indicate sliding friction intervals. |
|
Rack-and-pinion model. The unexpected minima of Ea(Rs) can be explained by simple engineering – a "rack-and-pinion" model. Consider the molecule as a cogwheel (the pinion) with L cogs, primitive radius Rm and external radius R*= Rm+ h, where h ∝ Rsl. The chord distance between nearest cogs is Rll*= 2R*sin(π/L). Best rolling conditions are expected when Rll* matches the substrate potential period Rs, i.e., for Rs(1)= Rll* and its fractions, Rs(2)= Rll*/2, Rs(3)= Rll*/3, etc. The main minimum of Ea(Rs) is expected at
|
(1) |

As shown in Fig.4, the cogwheel model, Eq.(1) with h=βRsl, where β is a parameter, fits extremely well the shift of minimum position with molecular size L. Moreover, it can explain its variation with load (the radius R* and therefore h decreases as the load grows) as well as with the lubricant-substrate interaction Vsl (R* and h decreases with Vsl).
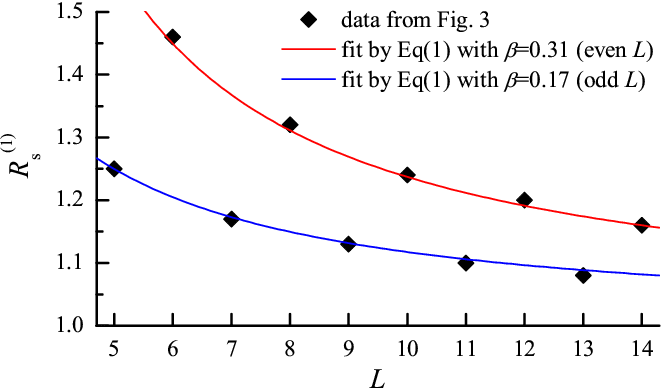 |
Figure 4: Position Rs(1) of the main minimum of Ea(Rs), extracted from Fig.3, for increasing molecular size L. Curves are fits to the cogwheel model Eq.(1). The asymptotic limit of 1 is still relatively far for reasonable molecular radii. |
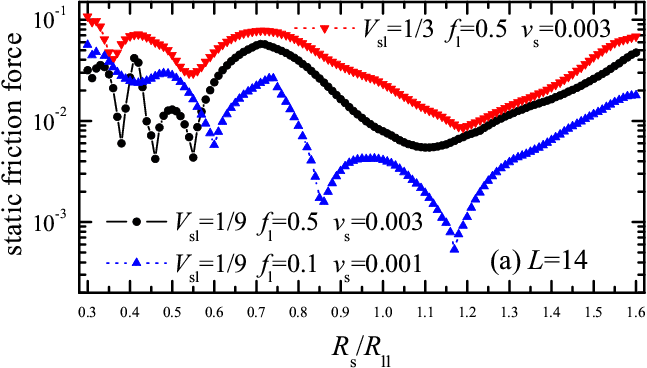
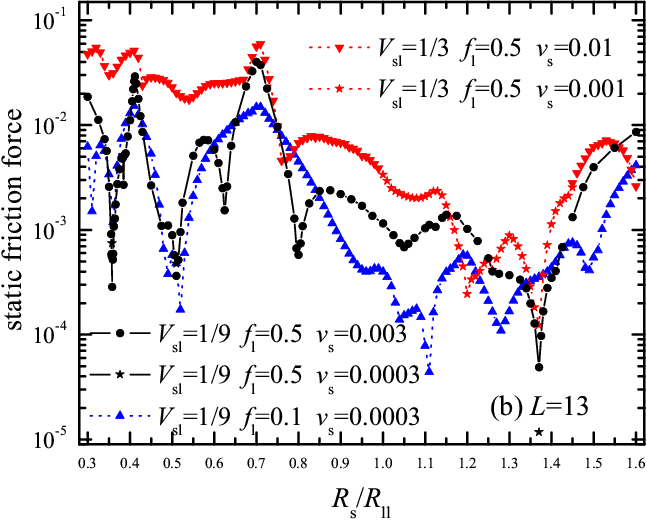
Simulation. These insights for rigid molecules are confirmed by the static friction force obtained from simulation with deformable molecules (see Fig.5). The friction coefficient μs= fs/fl ranges from μs~ 0.1 at Rs/Rll≈ 0.7 to μs~ 0.01 or even μs~ 0.001 at Rs/Rll ≈ 1.1. The results are also robust to changes of model parameters. For example, Fig.5 compares the dependences fs(Rs) for two values of the amplitude of lubricant-substrate interaction, Vsl=1/9 and 1/3, and for two values of the load, fl= 0.5 and 0.1. We found both static fs and kinetic friction fk to increase approximately linearly with load, fs,k ≈ f0s,0k+ μs,k fl. Visualization of MD trajectories shows that for Rs/Rll= 0.7, where friction is high, rolling rotation is in fact discontinuous, and accompanied by a molecular shift/sliding – much as cogwheels with excessive clearance would do – which dissipates mechanical energy into vibrations, whereas for Rs/Rll= 1.1, where friction is low, the motion is a smooth rotation, corresponding to optimal rack-and-pinion coupling (movies can be found here 6.2 Mb).
 |
 |
|
Figure 5: Static friction force fs as a function of the substrate lattice constant Rs/Rll for (a) L=14 and (b) L=13 for different system parameters: (i) fl = 0.5 and Vsl= 1/9 (solid curve and circles and stars), (ii) fl = 0.5 and Vsl= 1/3 (down triangles and red dotted curve and stars), and (iii) fl = 0.1 and Vsl= 1/9 (up triangles and blue dotted curve). Other parameters are: Rsl= Rs, Km= 100 and Vll= 1. |
|
Simulations further showed that this scenario remains valid at nonzero temperature T. As T increases, both static and kinetic friction forces are found to decrease, the stick-slip changing to creep and finally to smooth motion at high temperatures. We also found transitions from stick-slip to smooth rolling for increasing velocity. The cogwheel effect remains, and the calculated static friction for Rs/Rll= 0.7 and Rs/Rll= 1.1 still differ by a factor of ten or more. The critical velocity vc of the transition from stick-slip to smooth rolling also differs by a factor of about four in the two cases. In all cases we find fk << fs.
The present approach to the single rolling molecule can be extended to a finite coverage of lubricant molecules. For M molecules the individual molecular load is fl1= fl /M so that the friction per molecule fs1,k1 also decreases. However, the total friction force fs,k= Mfs1,k1 increases, so that the combined effect is a slow increase of the total friction with M. In a real situation, coalescence may lead to jamming, with molecules blocking their mutual rotation. In our model, jamming starts already at ΘM ≈ 0.1 and completely destroys rolling at ΘM > 0.3 (here ΘM = M/M1 is the coverage, with M1 the number of molecules in a monolayer).
Rolling spherical lubricant molecules can indeed provide better tribological parameters than sliding atomic lubricants. The effect may be as large as in macroscopic friction, where rolling reduces friction by a factor of 102 − 103, however only for low concentration of lubricant molecules, and for specially chosen values of the ratio Rs/R0, e.g., for Rs/R0 ~ 1.1, corresponding to perfect rack-and-pinion matching.
To conclude, rolling friction with a lubricant made of spherical molecules is effective if:
the substrates are more rigid than the lubricant, and the substrates (or protecting coverages) are specially chosen:
inert nonmetal surface (e.g., self-assembled monolayers) could represent a better choice than metals for fullerene deposition
increasing load could offer the simplest tool to change the rack-and-pinion matching through a pressure-induced decrease of h = βRsl
there are no jams; ways to avoid jams may be the following:
to decrease the concentration of lubricant molecules (θ < 1)
to increase temperature in order to break intermolecular bonds within the lubricant
to use charged lubricant molecules (?)
See the articles (pdf files may be found here):
O.M. Braun, Phys. Rev. Lett. 95 (2005) 126104 "Simple model of microscopic rolling friction"
O.M. Braun and E. Tosatti, Phil. Mag. Lett. 88 (2008) 509 "Rack-and-pinion effects in molecular rolling friction"
O.M. Braun and E. Tosatti, J. Phys.: Condens. Matter 20 (2008) 354007 (2008) 509 "Molecular rolling friction: the cogwheel model"
Last updated on November 26, 2009 by O.Braun. Copyright © by O.Braun. Translated from LATEX by TTH